RNA editing refers to post-transcriptional processes that modify mature RNA sequences relative to their genomic DNA template. In animals, the best-characterized events are A-to-I substitutions mediated by ADAR enzymes and C-to-U substitutions mediated by APOBEC enzymes. These modifications can diversify transcript sequences, alter coding potential, affect RNA stability and contribute to gene regulation.
The rise of high-throughput sequencing made transcriptome-wide RNA editing screens possible, but it also exposed a difficult methodological problem. Apparent RNA-DNA differences can arise from true editing, inherited polymorphisms, sequencing errors, mapping artifacts, splice junction misalignment, paralogous regions or incomplete reference assemblies. During my PhD, the field was marked by strong discrepancies between studies, with estimates ranging from a few dozen to millions of edited sites depending on filtering strategies and experimental design.
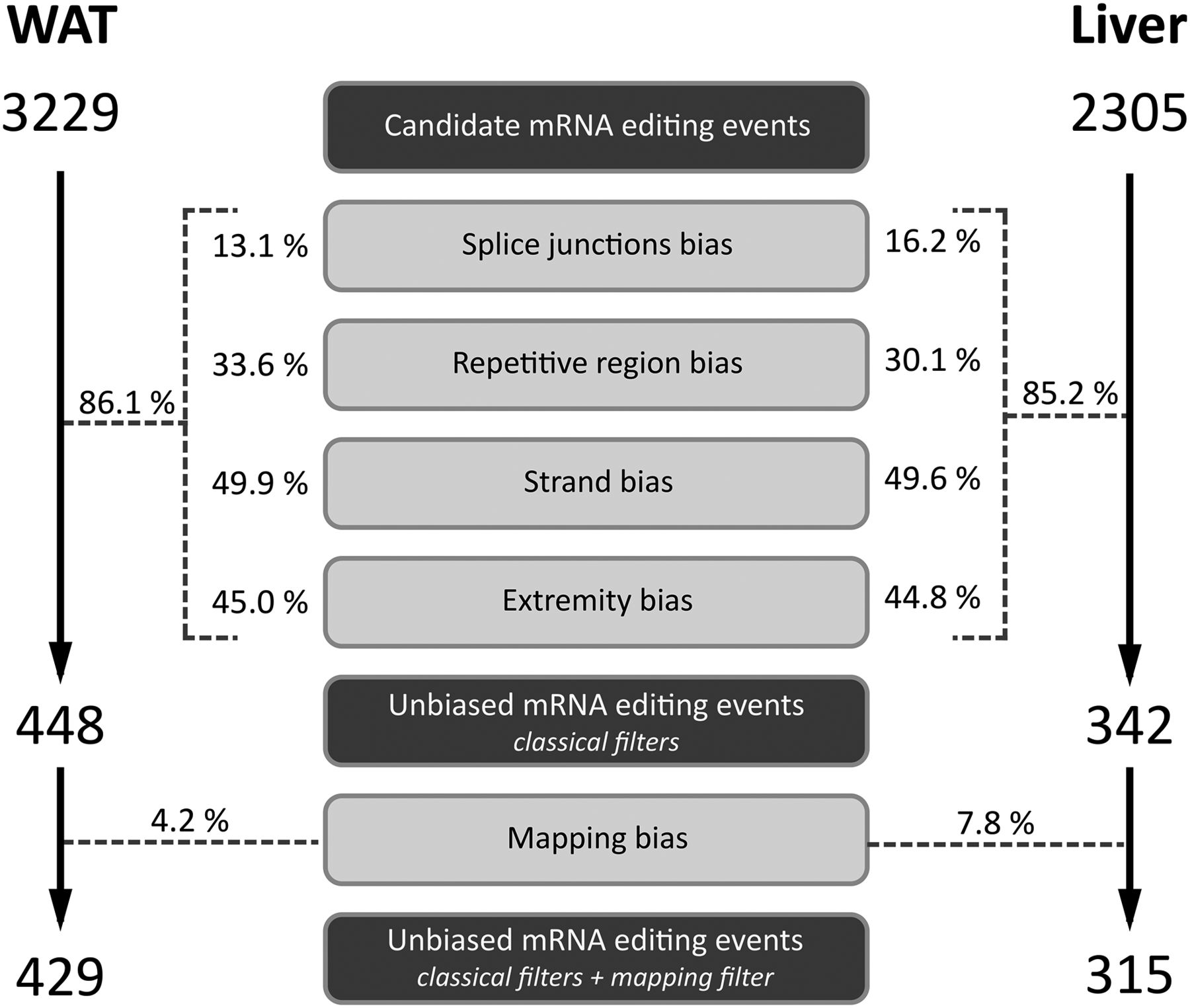
To address this, I developed a stringent WGS/RNA-seq strategy for calling transcript-specific editing events while explicitly controlling for technical artifacts and biological replication. The core principle was simple: a candidate event should survive comparison to matched genomic DNA, sequencing quality filters, splice-aware mapping checks, local sequence context controls, and additional tests against unmapped genomic sequences to account for gaps or errors in the reference genome.
Applied to chicken liver, adipose tissue and embryos, this framework revealed that the number of high-confidence mRNA editing events is limited in this species, with fewer than 20 robust sites per tissue under stringent criteria. Several events were conserved with mammals, supporting the idea that core RNA editing mechanisms predate the sauropsid-synapsid divergence. The project also showed that editing levels can vary with tissue, genotype, age, diet and sex, linking RNA editing to biological context rather than treating it as a purely static sequence feature.
Beyond the biological findings, this project trained my approach to genomics: rigorous artifact control, matched DNA/RNA designs, biological replication, and explicit interpretation of uncertainty are essential when extracting biological meaning from high-throughput sequencing data.