Cellular senescence is a stress response triggered by telomere dysfunction, oncogene activation, DNA damage, oxidative stress and other insults. It leads to a durable proliferative arrest, but also to profound changes in chromatin organization, transcription, metabolism and secretory activity. This dual nature makes senescence biologically fascinating: it contributes to tumor suppression, wound healing and development, yet persistent senescent cells can also promote chronic inflammation, tissue dysfunction, cancer progression and aging-related pathologies.
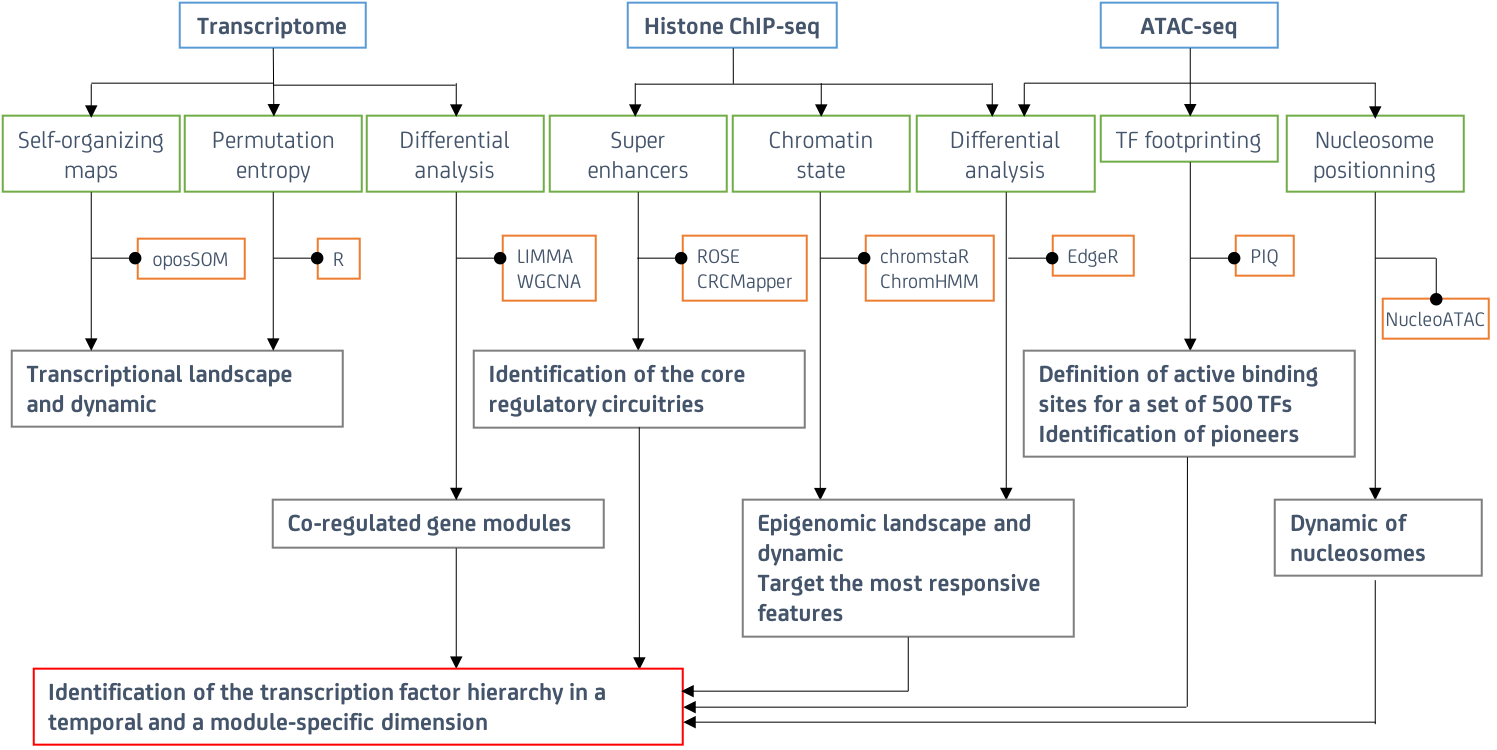
During my postdoctoral work in Oliver Bischof’s group at Institut Pasteur, I addressed a central question: how do diverse upstream stresses converge into a stable senescent cell fate? To answer this, I implemented time-resolved multi-omic profiling of primary human fibroblasts undergoing oncogene-induced, replicative and stress-induced senescence. The project combined RNA-seq, ChIP-seq for active and repressive chromatin marks, ATAC-seq, and metabolomics to reconstruct the temporal sequence of molecular events that establish and maintain the senescence program.
The analytical challenge was to transform high-dimensional temporal datasets into a mechanistic model of cell fate control. I developed integrative workflows using self-organizing maps, permutation entropy, chromatin state analysis, digital footprinting and network reconstruction to connect enhancer remodeling with transcription factor activity. These analyses revealed that senescence-associated transcriptional changes are largely encoded at distal regulatory elements rather than promoters, and identified AP-1 family members as pioneer-like hubs organizing the enhancer landscape of senescent cells.
This work provided a dynamic view of senescence as a regulatory trajectory rather than a static endpoint. Functional perturbation of cJUN was sufficient to reverse several features of oncogene-induced senescence in vitro, including proliferative arrest and SASP gene expression, highlighting AP-1-centered circuits as potential senomorphic or senolytic targets. The same integrative framework later contributed to studies of senescence escape, chromatin memory and cancer progression.
This project shaped much of my current research program: understanding how metabolic states, chromatin organization and transcription factor networks cooperate to control cell fate decisions in aging and cancer.