
The ultimate goal of quantitative trait loci (QTL) mapping is not only to locate genomic regions associated with a phenotype, but to identify the genes and mechanisms that shape complex traits. This remains difficult because QTL intervals often span megabases and contain many positional candidates. Without additional information, causal inference tends to favor already-known functional candidates, which narrows discovery and can miss unexpected regulators.
During my PhD, I worked on the genetic architecture of adiposity and energy metabolism in chicken models. In collaboration with Simon Boitard, I developed an integrative strategy combining classical linkage analysis, whole-genome sequencing, selection signature mapping with hapFLK1, coding variant annotation and cis-eQTL analysis. The aim was to progressively reduce large QTL intervals into prioritized candidate genes without relying only on prior biological assumptions.
This framework proved useful in divergent chicken lines selected for abdominal fat deposition. By overlaying QTLs with selection signatures and expression regulation, we refined candidate regions and identified PRKN and JAG2 as strong candidate regulators of adiposity. The results were strengthened by functional annotation of variants, allele-specific expression analyses and collaborative validation in mammalian models.
Conceptually, this project illustrates a strategy I still use today: combine complementary molecular layers to move from association to mechanism. Instead of treating genome-wide signals, expression variation and functional annotation as separate analyses, the project integrated them into a single prioritization framework for complex phenotypes.
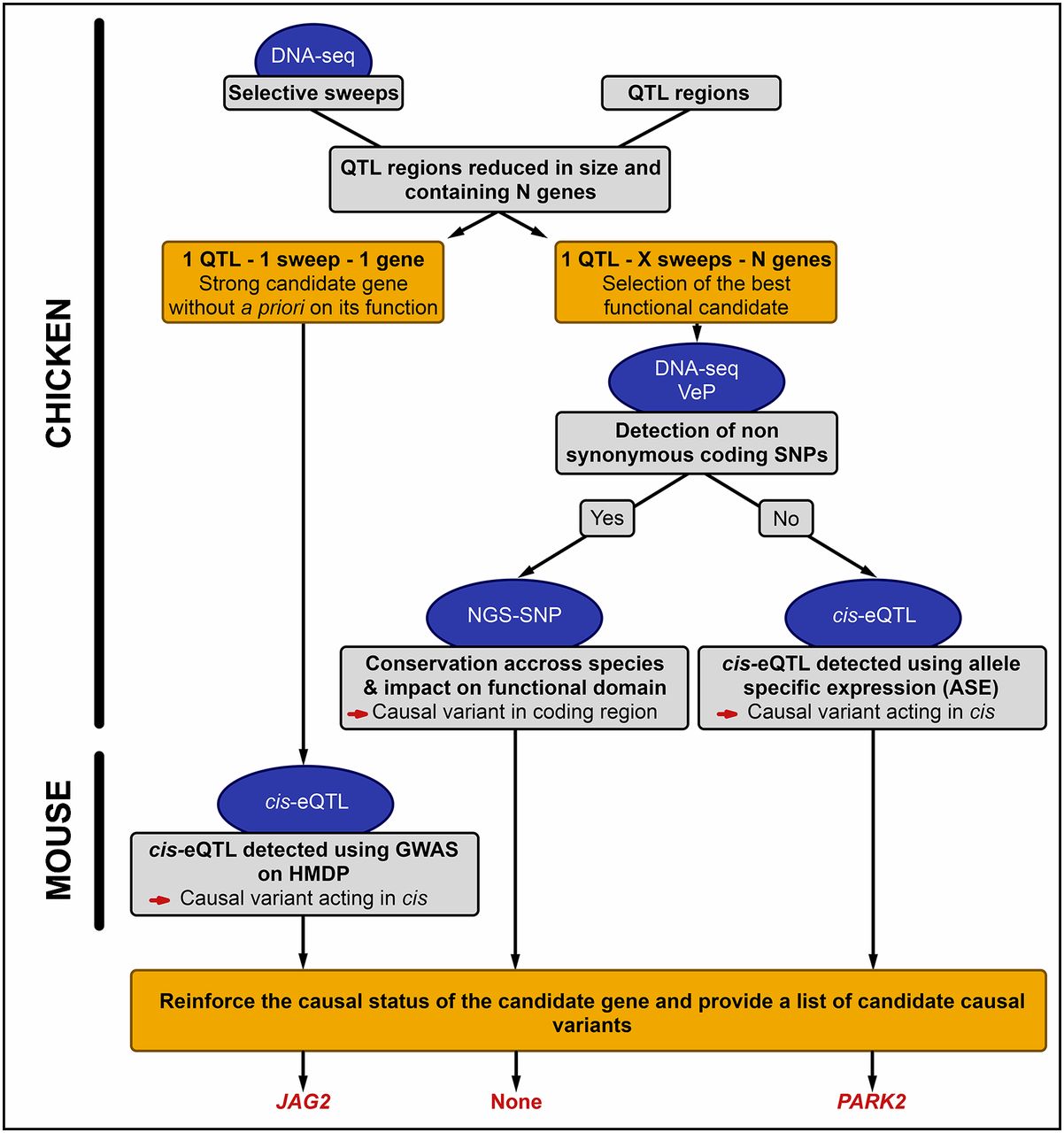
Overview of the strategy used to combine WGS and QTL mapping. QTL: quantitative trait locus; cis-eQTL: QTL for gene expression and acting in cis; ASE: allele-specific expression; VeP: Variant Effect Predictor tool from the Ensembl API; NGS-SNP: variant annotation tool; GWAS: genome-wide association study; HMDP: Hybrid Mouse Diversity Panel.
- For more details on hapFLK method, please refer to Fariello et al. 2013 ^